 |
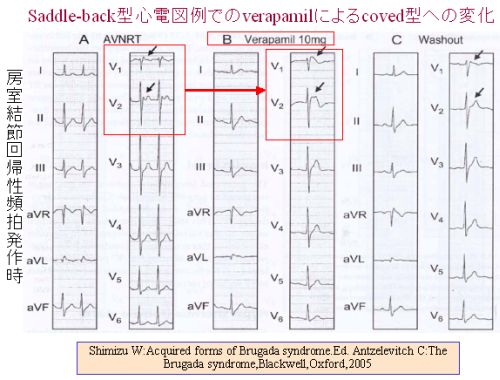
| 房室回帰性頻拍発作に対してCa++拮抗薬(ベラパミル)静注を行ったところ、頻拍発作は 停止したが,V1,2の心室群波形が典型的なcoved型Brugada心電図波形を示した。ベラパミル のwash out後には心電図は正常波形に復している。 |
Brugada症候群(15)
| トップページへ | Brugada(16)へ | Brugada(14)へ |
Brugada症候群における遺伝子異常
Brugadaらの最初の経験例は、1986年に彼らの外来を受診したポーランド人の3歳の男児でした。この患児は頻発する意識喪失発作を起こしていましたが、そのつど父が蘇生法を実施することにより回復していました。
本例の妹は、2歳の時に頻発する意識喪失発作の後に突然死しています。このような例をみると、Brugada症候群が遺伝性疾患であることは誰もが考えることです。しかし、当初は未だ本症候群の遺伝子異常は明らかでありませんでした。
1.ChenらによるSCBN5A変異遺伝子の発見:
1998年、Chenらは家族的に認められたBrugada症候群において、心筋細胞膜のNa+チャネルのαサブユニットをコードする遺伝子SCN5Aに変異があることを見い出しました。そのため、Brugada症候群は先天性QT延長症候群と同様に、分子の異常が致死的不整脈を惹起する一種の分子病であることが明らかとなり、広く注目を集めるようになりました。
2. Brugada症候群における遺伝子異常
Chenらにより発見されたSCN5A変位は、Brugada症候群の原因遺伝子の中で、最も頻度が高く重要な遺伝子ですが、このSCN5A変異はBrugada症候群の11-28%に認められるに過ぎません。したがって、Brugada症候群の発症に関与する遺伝子に関する研究が世界各地で行われ、現在は下表に示すように18種類の遺伝子変異が発見されており、今後も更に他の遺伝子変異が見いだされる可能性が強いと考えられます。
| 病型 | 責任遺伝子 | 障害される電流 | 頻度 |
| BrS1 | SCN5A | 内向きNa電流 | 11-28% |
| BrS2 | GPD1L | 内向きNa電流 | まれ |
| BrS3 | CACNA1C | L型Ca電流 | 1.6-1.9% |
| BrS4 | CACNB2 | L型Ca電流 | |
| BrS5 | SCN1B | 内向きNa電流 | まれ |
| BrS6 | SCNE3 | 一過性外向きK電流 | まれ |
| BrS7 | SCN3B | 内向きNa電流減少 | まれ |
| BrS8 | KCNH2 | 急速活性化遅延整流K電流 | まれ |
| BrS9 | KCNJ8 | ATP感受性K電流 | まれ |
| BrS10 | CACNA2D1 | L型Ca電流 | まれ |
| BrS11 | MOG1 | 内向きNa電流 | まれ |
| BrS12 | KCNE5 | 一過性外向きK電流 | まれ |
| BrS13 | KCND3 | 一過性外向きK電流 | まれ |
| BrS14 | HCN4 | ペースメーカー電流 | まれ |
| BrS15 | SLMAP | 内向きNa電流 | まれ |
| BrS16 | TRPM4 | Ca活性化非選択的陽イオン電流 | まれ |
| BrS17 | SCN2B | 内向きNa電流 | まれ |
| BrS18 | FGF12 | 内向きNa電流 | まれ |
(石川泰輔、蒔田直昌:Brugada症候群の遺伝子診断.池田隆徳ら編:不整脈症候群、南江堂、東京、2015から改変引用)
3. GPD1L
2006年、LondonらはBrugada症候群の大家系からGPD1L(glyserol-3 phosphate dehydrogenase like gene)を新しいBrugada症候群の責任遺伝子として同定しました。この遺伝子に障害があるとNa+チャネル蛋白の移送障害を起こしてその機能障害を起こします。
4.AntzelevitchらによるL型Ca++チャネル遺伝子変異の発見
1) Ca++拮抗薬によるBrugada 型心電図の顕性化
臨床的にCa++拮抗薬によりBrugada型心電図の顕性化が起こることが知られており、Ca++チャネルをコードする遺伝子変異とBrugada症候群との関連について興味が持たれていました。
Shimizuは、saddle-back型Brugada心電図を示す房室結節回帰性頻拍例の頻拍停止を目的としてCa++拮抗薬(ベラパミル10mg)を静注して 頻拍発作の停止に成功しましたが、その際、心電図波形はsaddle-back型からcoved型に変化し、ベラパミルのwash outにより再びsaddle-back型に復した例を報告しています。このような例の存在は、Brugada型心電図の形成にCa++チャネルの関与があることを示しています。
|
| 房室回帰性頻拍発作に対してCa++拮抗薬(ベラパミル)静注を行ったところ、頻拍発作は 停止したが,V1,2の心室群波形が典型的なcoved型Brugada心電図波形を示した。ベラパミル のwash out後には心電図は正常波形に復している。 |
2) CACNA1C およびCACNB2変異によるBrugada症候群
2007年、Antzelevitichらは、Brugada症候群の発端者連続82人について臨床的および遺伝学的検討を行い、7例(8.5%)にL型Ca++チャネルをコードする遺伝子の機能喪失型(loss-of-function)変異を認めました。
興味深いことは、これらの7例中3例(42.9%、全発端者82例中の3.7%)にQT間隔短縮(shorter
than normal QT interval)を認めたことで、これらはBrugada症候群の中で特異な心電図所見を示す1つのサブグループを形成しています。
3) QT間隔短縮を示すBrugada症候群症例
Antzelevitchらは心臓L型Ca++チャネルの支配遺伝子であるCACNB12b変異またはCACNA1Cに変異を認めたBrugada症候群症例において次のような特徴的所見を認めました。
(1) Brugada症候群としての表現型(phenotype)を示す(臨床所見、心電図所見など)。
(2) QT間隔短縮(shorter than normal QT interval)を示す。
(3) 急死の家族歴がある。
これらの例では右側胸部誘導の著明なST上昇、J波の顕著化、そのⅠ群抗不整脈薬(アジマリンなど)による増強、高位右側胸部誘導心電図におけるBrugada型心電図の顕性化などのBrugada症候群の際の特徴的心電図所見を認め、失神などの臨床症状を示す例もあります。
一般に心拍数60/分に相当する予測QT間隔の平均値は410msecで、(平均-2x標準偏差)で示されるQT間隔の正常下界は360msecとなります。
2000年、GussakらがQT短縮症候群について報告して以来、QT間隔の正常下界についての関心が高まり、QT短縮症候群の診断基準としては<300msecという値が用いられるようになっています。
AntzelevitchらがCa++チャネルをコードする遺伝子変異例に認めたBrugada症候群の新しいサブグループにおいては、QT短縮症候群で見るような<300msecという著しいQT間隔の短縮を認めませんが、正常下界(男性:≦360msec、女性:≦370msec)よりも短いQT間隔を示すため、Antzelevitchらはshorter
than normal QT inervalという表現を用いています。Antzelevitchらの論文中に紹介されている本症候群に属する3例中の第1例を以下に紹介します。
症例:25歳、男性
主訴:心停止、蘇生例
家族歴:弟(23歳)は21歳時から失神発作がある。10例の家族について遺伝学的および臨床的検討を行い、6例がBrugada型心電図と共にQT間隔短縮を示し、このような表現型(phenotype)を示す全例でCACNB2b変異を認めた。
心電図所見:下図は本例の心電図です。QTc間隔は330msecと短縮しています。V1,2に小さいr'様の波(J波)がありますが、ST上昇は著明でなく、saddle-back型Brugada心電図と診断されます。 ajmaline静注後のV1-3の2肋間上方の記録(V1''-V3'')では、著しいJ波の増高、ST上昇、coved型ST-T波形の出現などの典型的なBrugada型心電図所見を認めます。本例ではICD植えこみが行われました。
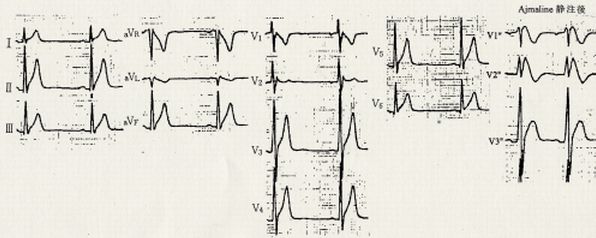 |
| CACNB2b変異を認めたBrugada症候群(BrS4)の心電図(説明上記) (Antzelevitch C, al: Circulation 115:442,2007) |
Antzelevitchらにより報告されたこのような例の存在は、Brugada症候群とQT短縮症候群との密接な関連を示しています。
4) SCN1B
Watanabeらは、SCN5A変異がないBrugada症候群282例と心臓伝導障害を有する44例の発端者について、心筋Na+チャネルのβサブユニットをコードする遺伝子SCN1Bについて検討し、この遺伝子の変異を有するBrugada症候群を同定しました。
5) KCNE3
Delponらは、K++チャネルのαサブユニットと協調してItoチャネルを構成するβサブユニットをコードするKCNE3変異によるBrugada症候群症例を見いだしました。この変異はItoを増加する機能亢進型変異です。
6)遺伝子多型(polymorohism)
ヒトの遺伝子は約22,000個あり、その塩基配列は個人により異なる部分があります。このような塩基配列の異常は、その人毎の病気に対する罹り易さ、薬の効き方、酒の強さ、顔の形などの個人差の決定に関与しています。
遺伝子変異というのは、そのような異常が集団の1%以下でしか認められない場合をいい、1%以上で認められる場合は遺伝子多型と呼びます。遺伝子多型の中で、塩基配列の異常が1か所のみで認められる場合を一塩基多型(SNPs,
スニップス, single nucleotide polymorphisma)といいます。
Brugada症候群の成因として、このような一塩基多型が関与している可能性が指摘されています。Bezzinaらは、Brugada症候群症例のプロモーター領域に6個の一塩基多型を認めており、このような遺伝子多型は日本人では25%の高率に認められますが、白人や黒人では全く認められないことを報告しています。
 Brugada症候群(16)へ
Brugada症候群(16)へ