 |
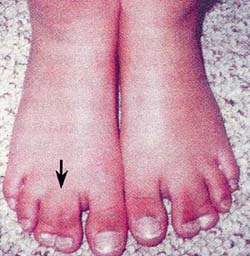 |
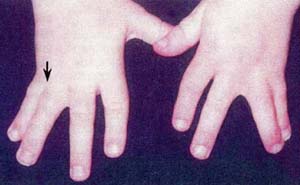
| 左右対称性を示す第4,5指の合指症 | 左右対照的な第2,3趾の合指症 |
| (Splawski I et al:Cell Vol.13,19-31,2004 から引用) | |
| 遺伝性不整脈へ | LQT目次へ |
| 二次性QT症候群へ | トップ頁へ |
1992年、Reichenbachらは、心臓興奮伝導障害と手足の合指(syndactyly)を合併し、生後5カ月で突然死した小児例について発表している(Kinderarztl, 60:34-36,1992)。1995年, Marksらは心電図上のQT間隔延長と合指(syndactyly)を持ち、急死の危険が極めて高い症例を2例について報告し(Marks
ML et al:JACC 1995; 25:59-64), これがQT先天性QT延長症候群の新しい型であることを発表した。
Splawski I, Timothy KWらは、類似した所見を示す17例(内男性9例、女性8例)に認められた種々の臨床症候の頻度を示すと共に、その遺伝子解析を行い、本症候群の原因遺伝子を明らかにすると共に、これらの一群の疾患を「Timothy症候群」と呼ぶことを提唱した。
我が国においても、福見らが1歳男児例を報告している(平成17年小児心電図研究会)。
1. Timothy症候群の症状および所見
Marksら(1995)の発表では、身体奇形としてはsyndactyly(合指)のみが強調されたが、その後の研究から本症候群は,次のような多様な症状・所見を示す他系統障害を起こす遺伝子異常であることが判明した。
1) 出現頻度が極めて高い症状・所見(100%近くに出現)
(1) 極めて著しいQT間隔延長、
(2) 合指(syndactyly):単純性合指で、左右対称的に、両側上・下肢に認めることが多い。
(3) 生直後からの禿頭。
(4) 小歯症(small teeth)
2) 上記よりは頻度が少ない症状・所見
(1) 心奇形(卵円孔開存、動脈管開存、心室中隔欠損、その他)
(2) 自閉症
(3) 知的発育障害
(4) 顔面形態異常
(5) 近視
(6) 免疫不全
(7) 間欠的低血糖
(8) 低体温
(9) 筋疾患
(10) 言語発育遅延
Splawskiらは、17例(男性9例、女性8例)の本症候群症例に見られた種々の臨床所見および身体徴候の頻度(%)を調査し、下表のようにまとめている。
| 症状、所見 | % | ||
| 心臓 | QT間隔延長 | 100 | |
| 不整脈 | 頻脈性心室性不整脈 | 71 | |
| 徐脈、房室ブロック | 94 | ||
| 先天性心奇形 | 動脈管開存 | 59 | |
| 卵円孔開存 | 29 | ||
| 心室中隔欠損 | 18 | ||
| ファロー四徴症 | 6 | ||
| 心肥大 | 35 | ||
| 中枢神経障害 | 自閉症 | 60 | |
| 自閉症系列の疾患 | 80 | ||
| 知能発育障害 | 25 | ||
| 痙攣 | 21 | ||
| 臍帯 | 2血管 | 13 | |
| 消化管 | gap reflex | 31 | |
| 皮膚 | syndactyly | 100 | |
| 生下時からの禿頭 | 100 | ||
| 顔面 | 形態異常 | 53 | |
| 眼 | 近視 | 25 | |
| 鼻 | 副鼻腔炎 | 29 | |
| 歯 | 小歯 | 100 | |
| 齲歯 | 50 | ||
| 肺 | 肺炎、気管支炎 | 47 | |
| 肺高血圧症 | 21 | ||
| 内分泌器官 | 甲状腺機能低下 | 8 | |
| 電解質、代謝 | 低Ca血症 | 33 | |
| 低血糖 | 36 | ||
| 低体温 | 33 | ||
| 筋肉 | 骨格筋低緊張 | 40 | |
| 免疫系 | 免疫不全、反復感染 | 43 | |
| (Splawski I et al:Cell Vol.13,19-31,2004 から改変引用) | |||
2.合指症 (syndactyly)
この所見は、Timothy症候群に極めて特異的な所見であり、本症候群のほぼ全例に認められており、合指症を認めた場合は本症候群を必ず考慮に入れなければならないとまで指摘されている重要な所見である。
一般的に、合指症は全出生の0.03%前後に認められる普遍的な先天性奇形で、常染色体性優性遺伝形式をとるものと劣性遺伝形式をとるものとがある。
合指症は次のように分類される。
1)単純性合指症(simple syndactyly):皮膚および軟部組織のみの融合がある例、
2)複雑性合指症(complex syndactyly):骨組織の融合がある例。
また融合の範囲(広さ)により完全型と不完全型に分ける。
1)完全型(compleete):distal phalanxにまで融合が及ぶ例、
2)不完全型(incomplete):proxysmal phalanxの中央を越えるが、distal
phalanxにまで融合が及ばない例。
合指は、左右両側に対称性に出現する例が多く(50%)、部位的には手の第3,4指間の合指例が最も多い。足ゆびの合指例もある。
下図に合指の実例を示す。。
|
|
| 左右対称性を示す第4,5指の合指症 | 左右対照的な第2,3趾の合指症 |
| (Splawski I et al:Cell Vol.13,19-31,2004 から引用) | |
3.特異な顔貌
Timothy症候群の患者は、特異な顔貌を示すことが知られている。
 |
| 丸い顔、扁平な鼻橋、上顎の退縮、薄い上唇などの特徴を示す。 (Splawski I et al:Cell Vol.13,19-31,2004 から引用) |
4.諸種の先天性心臓・大血管奇形の合併
卵円孔開存、動脈管開存、心室中隔欠損、ファロー四徴、血管輪、右側大動脈弓などの心血管異常をしばしば合併する。
5.心電図所見
1)著明なQT間隔延長
全例が極めて著明なQT間隔延長を示す。この著明なQT間隔延長が本症における致死的心室性不整脈の基盤となっている。
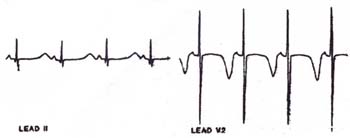 |
| QT間隔525msの著明なQT間隔延長を示す。 (Marks ML et al: JACC 1995;25:59-64) |
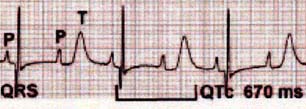
2)徐脈、房室ブロック
胎児期から著明な徐脈を認め、そのために心異常が気づかれる例も多い。この徐脈は洞徐脈によるが、房室ブロックもしばしば認められ、これが徐脈を助長している場合もある。しかし、この房室ブロックは房室伝導系の一次的な障害によるのではなく、下図に示すように著明なQT間隔延長のためであると考えられている。
 |
| (Splawski I et al: PANS 102:8089-8096,2005) |
3)T波の交互脈を示す場合もある(下図)。
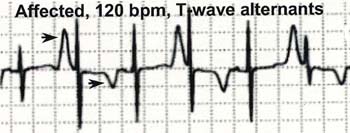 |
| (Splawski I et al:Cell Vol.13,19-31,2004 から引用) |
4)多形性心室頻拍、心室細動
本症候群の予後は重篤で、高率に多形性心室頻拍、心室細動を起こして急死する。
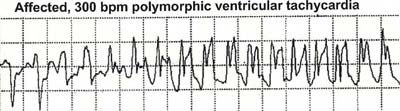 |
| (Splawski I et al:Cell Vol.13,19-31,2004) |
6.本症候群の成因
本症の成因は、心臓のL型Caチャネル(Cav1.2)をcodeする遺伝子CACNA1Cの単一変異により生じる。この変異は機能獲得型障害を起こし、心筋活動電流のうち、Ca電流を増強し(下図左)、そのため活動電位持続時間を延長させる(下図右)。このCACNA1C遺伝子変異は、心臓のみならず、身体の多くの臓器、組織に発現するため、本症患者は多様な身体所見を示す。
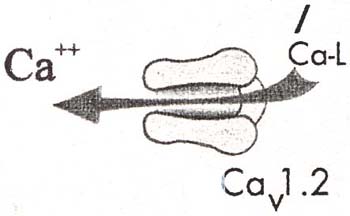 |
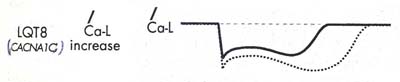 |
| 心筋L型Caチャネル | Ca電流増加による心筋活動電位持続時間延長 |
| Wilde AAM & Bezzina CR:Heart 2003;91:1352-1358)より改変引用 | |
下図は、本症候群において、Ca電流の増大が、心筋細胞内電位持続時間の延長を起こし、これが心電図のQT時間延長を起こす相互機序を説明する模型図である。
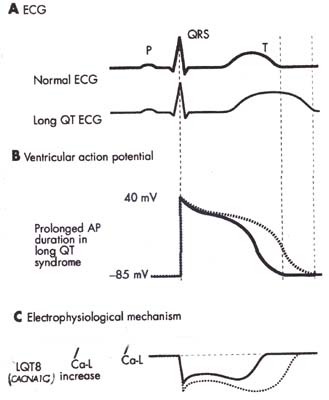 |
本症候群では、図Cに示すように、 心筋L型Caチャネル(Cav1.2)を codeする遺伝子CACNA1Cの 単一変異により、、内向きCa電流 が増加する。そのため、B図に示す ように心筋細胞活動電位持続時間 が延長する。その結果、A図に示す ように心電図の」QT間隔が延長する。 |
| (Wilde AAM & Bezzina CR:Heart 2003;91:1352-1358)より改変引用 | |
7.予後
本症候群の予後についての系統的研究は未だないが、著しく予後が悪く、発表された症例中、多くの例が突然死している。この突然死は著明なQT延長による多形性心室頻拍、torsade
de pointes, 心室細動に基づくものと考えられる。
8.治療
本症候群に特異的に有効な治療法は未だ認められていない。多くの例でβ遮断薬、メキシレチンなどの抗不整脈薬、あるいはペースメーカー植え込み療法が行われているが、非常に有効であるとは言い難い。植え込み型除細動器のimplantationの有効性が期待されるが、本症患者の多くが新生児、乳幼児であるために一般的治療法であるとは言い難い。頸部交感神経節切除術が一部の例で行われているが、必ずしも有効とは考え難い。